
Download Data from NCBI
If a research paper includes the analysis of sequence data, nearly all journals require that the raw data be deposited in a publically accessible repository. For example, if we look at the publication below:

in the Methods section we find the following:

Let’s download the 16S rRNA sequence data for this publication to use for our tutorial. We can do this with NCBI’s entrez-direct and SRA-Tools following the instructions given on the Blog page Downloading Sequences from NCBI’s SRA.
If you need to install entrez-direct and the SRA Tools you can do it with one of the environment managers. For instructions, see here. In the next code block I am using micromamba to install SRA Tools in an environment named sra.
micromamba create --name sra -c bioconda entrez-direct sra-toolsOpen your terminal and enter the following code, one line at a time:
# Create a directory for this project:
cd
mkdir example
cd example
# Activate the environment containing the SRA Tools
micromamba activate sra
# Get the run information for the project
esearch -db sra -query "PRJNA628034" | efetch -format runinfo > file_list.txt
# Extract a list of the accession IDs for the project
sed '1 d' file_list.txt | cut -d "," -f1 > sra_list
# From that list, extract a random sample of 6 accession IDs
shuf -n 6 sra_list > short_list
# Download the sequence data for those 6 accession IDs
cat short_list | xargs fastq-dump --split-files
# List the directory contents
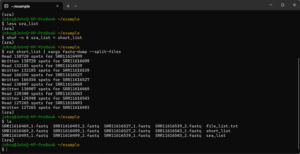
lsYou should see something like the following in your terminal. The sequence file names will be different because you drew a random sample of 6 samples from the total of 96. The number of “spots” is the number of sequences per file. Notice that there are separate files for forward and reverse reads as indicated by the digit just before the fastq file extension.
Sample the Sequence Files with SeqKit
There are more than 100,000 sequences in all of the sequence files I downloaded. To develop a workflow that can be quickly run on a personal computer we need to down sample the reads to 10,000 or so per file. We can do this with SeqKit. See the blog I wrote for how to install it. Make sure you do not have any other environment active before you create one for SeqKit.
If necessary, deactivate an environment not containing SeqKit and activate one that does.
micromamba deactivatemicromamba activate seqkit
Open an empty file named sample_seqs.sh with the nano editor.
nano sample_seqs.shCopy and paste the following code into the editor.
#!/bin/bashrm -rf sampled/mkdir sampled/for f in $(ls *.fastq); doout=${f/_/_R}seqkit sample $f -n 10000 -s 913 -o $outdonemv *_R?.fastq sampled/
Close the editor with Ctrl O, hit Return to save the file with the same name, Ctrl X to exit.
Make the bash script you just created executable and run it by entering the following:
chmod u+x sample_seqs.sh
./sample_seqs.shBecause we used the same seed (913) to sample all of the files, pairing of reads is maintained between the forward and reverse reads for each sample. This is very important for further analysis. Sampled files contaning 10,000 sequences each are written to the sub-directory sampled/. You can list them with:
ls -l sampled/Exercise
Choose a publication based on analysis of 16S rRNA gene sequences and determine the project accession ID under which sequence data were deposited. Determine the primers used and the variable region sequenced. Randomly choose six run IDs for the project and download 10,000 paired sequences for each run (sample).