
Introduction
There are many programs for merging forward and reverse reads. If you plan to use the QIIME2 DADA2 plug-in, you should allow it to merge the reads. It will correct errors in the reads prior to merging them. If you use other methods such as the QIIME 2 deblur plug-in or some other clustering method, you will need to merge the reads. There are instructions for using PandaSeq(installation instructions here and usage here) to merge reads elsewhere on this web site. QIIME 2 also includes vsearch which may be used natively or through the plug-in, but I prefer to use USEARCH. The 64-bit version is now free and its function fastq_mergepairs has the advantage that it can automatically remove overhangs (trim staggered pairs). It is also easy to use in an “investigative” manner to choose parameter settings. Unfortunately, there is no version for newer Apple computers (but the linux version could be run on an Apple computer via Docker or Ubuntu Box).
USEARCH
To install USEARCH (Windows or linux), download the appropriate binary from GitHub and place it on your path, shortening the name to usearch11 when you do so. If using Windows WSL2, install it there as for the linux version. The OSX versions are for Intel based Apple computers. If you have a newer (M series) Apple computer, you can install the linux version in a Docker or Virtual Box linux environment.
Investigative Mode
Open your terminal and move into the directory containing the sequences you wish to merge. Then enter the following, using a forward file name in your directory.
usearch11 -fastq_mergepairs SRR11616469_16S_R1.fastqYou do not have to give the name of the reverse file if the forward file name includes “R1.” The reverse file will automatically be found. And you do not have to give an output file. None will be written. If the R1 convention is not followed, give the name of the reverse file with -reverse <reverse file name>.
This immediately gives a lot of information.
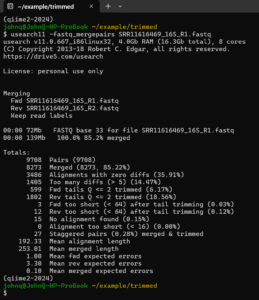
Most of the pairs that did not merge had too many differences (more than the default value 5) in the overlap region (192 bp). Given such a long overlap region, a limit of 5 differences is very restrictive. We can allow more (10) differences with -fastq_maxdiffs 10 :
usearch11 -fastq_mergepairs SRR11616469_16S_R1.fastq -fastq_maxdiffs 10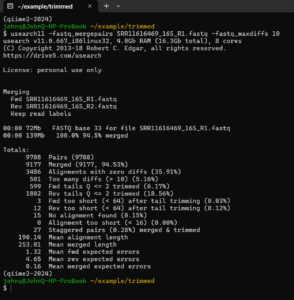
Now more than 94% of the sequences merge and the mean expected error for the merged sequences is only 0.16. To have the result written to an output file, give -fastq_out <file name>.
USEARCH Script
Below is a bash script for merging all read pairs in a directory. In addition to the parameters used above, it filters/truncates all merged sequences to a length of 253 bp, the expected amplicon length for the V4 region.
#!/bin/bash
cd ~/example # Move to project directory.
# Remove previous results
rm merg_log.txt
rm -rf merged
mkdir merged
# Move into directory containing reads to be merged
cd trimmed
# Merge all read pairs with usearch.
# File names are assumed to follow _R1/_R2 convention.
# Otherwise edit as warranted.
# Edit maximum differences allowed as warranted.
for f in $(ls *_R1.fastq)
do
usearch11 -fastq_mergepairs $f -reverse ${f/_R1/_R2} \
-fastq_maxdiffs 10 \
-fastqout ../merged/${f/_R1/_merged} \
-fastq_maxmergelen 253 -fastq_minmergelen 253
doneVSEARCH
Below is a comparable bash script for using VSEARCH to merge all paired sequence files in a directory. VSEARCH is part of QIIME 2, so the script can be run with an activated QIIME 2 environment. The drawback to using VSEARCH is that overhangs are not automatically trimmed, so you have to be careful when trimming the sequences that you eliminate the possibility of overhangs when merging.
#!/bin/bash
cd ~/example # Move to project directory.
# Remove previous results
rm -rf merged
mkdir merged
# Move into directory containing reads to be merged
cd trimmed
# Merge all read pairs with usearch.
# File names are assumed to follow _R1/_R2 convention.
# Otherwise edit as warranted.
# Edit maximum differences allowed as warranted.
for f in $(ls *_R1.fastq)
do
vsearch -fastq_mergepairs $f -reverse ${f/_R1/_R2} \
-fastq_maxdiffs 10 \
-fastq_allowmergestagger \
-fastqout ../merged/${f/_R1/_merged} \
-fastq_maxmergelen 253 -fastq_minmergelen 253
done