
Introduction
We will ultimately use QIIME 2 to generate tables giving the number of counts by sample for each OTU (Operational Taxononomic Unit) or ASV (Amplicon Sequence Variant), but there are several things we may (or wish) to do before getting to that stage. Depending on exactly how the sequencing was done and what was returned by the sequencing faciltiy, we may have to do the following:
-
-
- Demultiplex reads (sort by sample)
- Split interleaved reads
- Check read quality
- Trim primers from reads
- Merge paired reads
-
To know which of these steps are actually necessary, you have to know certain things about the raw data. That is, you have to know your sequences.
There are several sources for the required information. Depending on how much preparation you had to do before submitting samples to the sequencing faciltiy, you may already know somethings. If you submitted amplicons, then you know what primers were used and therefor what gene region was targeted. If your primers included barcodes, you know the barcode for each sample. Some of the information you can learn by inspection. Commercial sequening facilities usually return the required information with the data or provide it on their website. If the sequencing was done by a facility at your institurion, you may have to ask.
For the data used in our tutorial, we can get the required information from the publication and by inspection.
Demultiplexing
Demultiplexing refers to sorting the reads by sample. If this has already been done for you, you will have one or two (if reads are paired) files per sample. Otherwise you will need to sort the reads using a file associating barcodes present in each read with a sample. This can be done in QIIME 2 and an example is provided in the Moving Pictures tutorial on the QIIME 2 website..
For the data used in our tutorial, we recognize that demultiplexing is not necessary. We have one forward and one reverse sequence file for each sample.
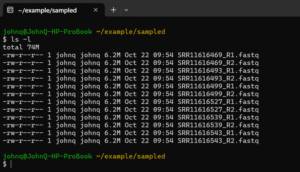
But in this case the file names are meaningless to us. We have to associate them with a sample name. We will do this using a manifest file when we import the data into QIIME 2. We can get the associations from the file_list.txt file we downloaded for the project.
Splitting Interleaved Reads
In some cases forward and reverse reads are in the same file. I this is the case, inspecting the file gives something like this:
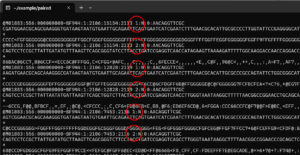
The number 1 in the circles indicates a forward read and the number 2 indicates a reverse read. To import into almost all programs (including QIIME 2) the forward and reverse reads need to be split into separate files. This can be accomplished with SeqKit’s split2 command. If the following code is run from the directory containing interleaved_file.fastq, separate files for forward and reverse reads are written to the sub-directory split_files as interleaved_file.part_001.fastq and interleaved_file.part_002.fastq.
mkdir split_filesseqkit split2 -p 2 interleaved_file.fastq -O split_files
Check Read Quality
I will cover how to check read quality on the next page, but I want to mention two potential problems here that may influence your workflow. It is typical for read quality to decline with read length, especially for the reverse reads. If severe enough this may reduce the overlap between quality filtered forward and reverse reads to the point that merging is impractical. Also, there may be so few reads for some samples that they will have to be excluded from further analysis.
Trim Primers
If primers are present, they should be trimmed. I view this in part as a quality control step: if the primers are supposed to be there and they are not, then the reads should be discarded. But also, the portions of the sequences matching the primers are not informative, because they match the primers and not necessarily the sequence they amplified. This is obviously true if the primers are degenerate, but it is also possible to get some amplification when primers do not exactly match the sequence being amplified.
Binned Quality Scores
Some sequencing platforms output binned quality scores. Rather than having all possible quality scores encoded in the fourth line of the fastq files, for example from 1 to 40 in the case of MiSeq, there are only three or four. This is true of newer Illumina platforms (e.g. NovaSeq 6000, NovaSeq X/X Plus, NextSeq 1000/2000, iSeq 100), PacBio and Loop-Seq. This can have consequences for processing, especially with DADA2 which uses Q-scores to estimate transition rates from the Q-scores in order to correct sequencing errors.
Merge Paired Reads
Merging forward and reverse reads is advantageous because it gives longer sequences, making it possible to cover more than just the 16S rRNA gene V4 region: both V3 and V4 or V4 and V5, for instance. This should enable more accurate classification of the sequences. Merging can be done along with other steps with the QIIME 2 DADA2 plugin, but I sometimes get better overall read retention by merging first with USEARCH. I do not understand why, but Robert Edgar claims that USEARCH is the only program that correctly calculates Q scores in the overlap region. Maybe this is why?