
Introduction
As explained on the Know Your Sequences page, if primers are present they should be removed for two reasons: as a quality control step and to remove parts of the read that do not necessarily match the sequence amplified. In order to remove (trim) the primers, we obviously need to know their sequences. We also need to know the region they target and the expected length of that region so that we can evaluate trimming success.
You can get the primer sequences from several sources: from personal knowledge if you did the amplification, from the sequencing facility, or, in our example, from the publication. Li et. al., 2021 states they used primers 515f and 806rb as presented in Apprill et al., 2015. These primers target the V4 region (approximately 253 bp in length) of the 16S rRNA gene. Thus we have:
>515f
GTGCCAGCMGCCGCGGTAA
>806rb
GCACTACNVGGGTWTCTAATMapping Primer to Sequences
I always like to check the alignment of the primers to the reads. The first sequence in each of paired fastq files can be extracted as fasta sequences with a little bash.
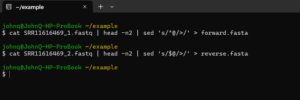
I then align the primers to these fasta sequences with BioEdit (Windows only) or MEGA (Windows, Mac OS or Linux). For our example we get:
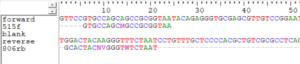
We see that both primers hit, but neither at the very beginnings of the sequences. The forward primer hits beginning at the sixth position and the reverse primer hits beginning at the second position. We need to take these facts into account in trimming the primers. We cannot “anchor” the primers (require that they be at the very ends of the sequences). See the cutadapt manual for detailed information about running the program.
Cutadapt is included in QIIME 2, and with an active QIIME 2 environment we can trim the primers with cutadapt either natively or using the QIIME2 plugin. For developing our workflow I will use cutadapt natively because one of the parameters I wish to use (-l) is not accessable via the plug-in. Also, I avoid having to import sequences into QIIME 2 and exporting the results.
Activate a QIIME 2 environment and run the following bash script.
#! /bin/bash
# Trim forward and reverse primers from paired reads.
# Input uncompressed files, output uncompressed files.
# The forward primer is 515f
fwd=GTGCCAGCMGCCGCGGTAA
# The reverse primer is 806rb
rev=GCACTACNVGGGTWTCTAAT
cd ~/example/ # Move into experiment level directory.
rm cutadapt_log.txt # Remove previous log file.
rm -rf trimmed # Remove previous results
mkdir trimmed # Make directory for the trimmed reads.
cd sampled/ # Move into sub-directory containing sampled reads.
# Trim using cutadapt.
# Force trimmed length to be 225 bp and format for input to Figaro.
for f in $(ls *_R1.fastq)
do
cutadapt -g $fwd -G $rev \
--discard-untrimmed \
-e 0.11 -O 15 -m 225 -l 225 \
-o ../trimmed/${f/_R1.fastq/_16S_R1.fastq} \
-p ../trimmed/${f/_R1.fastq/_16S_R2.fastq} \
$f ${f/_R1/_R2} >> ../cutadapt_log.txt
done
Explanations for the cutadapt arguments are given in the table below.
| Argument | Explanation |
| -g | The forward primer sequence |
| -G | The reverse primer sequence |
| –discard-untrimmed | If the neither primer is found the pair of reads is discarded |
| -e | Allowed mismatches are given as a fraction of the primer length; here floor(0.11 times 19) = 2 |
| -O | Minimum required overlap between primer and sequence |
| -m | Minimum trimmed length for retention; paired reads are discarded if less than 225 bp |
| -l | Maximum trimmed length; if longer, paired reads are truncated to 225 bp |
| -o | Path and file name for the trimmed forward reads; the output file name is modified to include “_16S”. |
| -p | Path and file name for the trimmed reverse reads: the output file name is modified to include “_16S”. |
| $f | Variable for the forward read file name, assigned in the for statement; input to cutadapt |
| ${f/_R1/_R2} | The reverse read, “_R2” is substituted for “_R1” in the forward read file name; input to cutadapt |
| >> cutadapt_log.txt | std (what is normally output to the screen as the program runs) is redirected to the file cutadapt_log.txt |
I modified the output file names to have three parts separated by underscores, e.g. SRR11616469_16S_R1.fastq and SRR11616469_16S_R2.fastq, and required both forward and reverse trimmed sequences to have the same (225 bp) lengths because I plan to use these trimmed sequences in a tutorial using the QIIME2 DADA2 plug-in with truncation parameters chosen using Figaro. Figaro requires file names to be formatted this way or the way Illumina formats file names. The lengths must be less than the expected amplicon length because DADA2’s merging step does not handle read-through leading to what Robert Edgar refers to as staggered pairs. This will become clearer below.
You can browse the log file with:
less cutadapt_file.txtThe log file records the parameters used to trim each file, the proportions of forward, reverse, and total reads passing the filters . It is useful in diagnosing problems if the results are not as expected.
You can also move into the trimmed directory and run the following script to count the number of sequences in each file and determine the length of the sequences in each.
#!/bin/bash
echo "file\t\tformat\ttype\tnum_seqs sum_len\tmin_len\tavg_len\tmax_len" > counts.txt
for f in $(ls *.fastq); do
seqkit stats $f | sed '1d' >> counts.txt
doneThen browse counts.txt with:
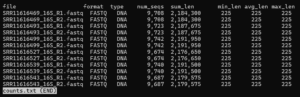
less counts.txtAfter trimming our sampled sequences, the number of sequences should be close to 10,000 (a few are lost because one or both primers were not found – quality control!) and all values in the last three columns should be 225 as below.
Read-through and Staggered Pairs
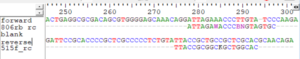
The sequences we downloaded from project PRJNA628034 (see page Sampling Data for Workflow Development) exhibit read-through leading to the staggered pairs problem because the primers target the 253 bp V4 region and the Illumina sequencing chemistry used produced 300 bp reads. The figure below shows how the reverse complement of the reverse primer hits near the end of the forward read and the reverse complement of the forward primer hits near the end of the reverse read. If we only trim the primers from the 5′-ends of the sequences, the trimmed reads will still be longer than the target region and merging will result in staggered pairs. We must prevent this in some way.
There are several solutions. One can force the trimmed sequences to be shorter as I did in the cutadapt script above. Another method would be to truncate all of the reads to be shorter (~ 250 bp in this case) before primer trimming. A third method is to merge using a program (USEARCH or VSEARCH) that can automatically remove overhangs. If processing with the R versiono of DADA2 the script allows for trimming overhangs when merging, and this option is now available with the QIIME2 DADA2 plugin as of the 2025.7 version. Still another method, one particularly suited to working with ITS data, is to modify the cutadapt script as in the section below.
Trimming ITS Sequences
The following cutadapt script eliminates the staggered pairs problem by trimming both ends of the sequences. The 5′-ends are trimmed as in the cutadapt script above, under the section Mapping Primer to Sequences. Additionally, the 3′-end of the forward reads are timmed using the reverse complement of the reverse primer, and the 3′-end of the reverse reads are trimmed using the reverse complement of the forward primer.
#! /bin/bash
# Trim forward and reverse primers from paired reads with read-through.
# Input uncompressed files, output uncompressed files.
# The forward primer is BITS
fwd=ACCTGCGGARGGATCA
# The reverse primer is B58S3
rev=GAGATCCRTTGYTRAAAGTT
# The reverse complements are:
fwd_rc=TGATCCYTCCGCAGGT
rev_rc=AACTTTYARCAAYGGATCTC
cd ~/example/ # Move into experiment level directory.
rm cutadapt_log.txt # Remove previous log file.
rm -rf trimmed # Remove previous results
mkdir trimmed # Make directory for the trimmed reads.
cd sampled/ # Move into directory containing sampled reads.
# Trim using cutadapt.
for f in $(ls *_R1.fastq)
do
cutadapt -g $fwd -a $rev_rc -G $rev -A $fwd_rc -n 2\
--discard-untrimmed \
-e 0.11 -O 15 --max-n 0 \
-o ../trimmed/${f/_R1.fastq/_16S_R1.fastq} \
-p ../trimmed/${f/_R1.fastq/_16S_R2.fastq} \
$f ${f/_R1/_R2} >> ../cutadapt_log.txt
doneThe additional arguments are explained in the table below:
| Argument | Explanation |
| -a | Reverse complement of reverse primer |
| -A | Reverse complement of forward primer |
| -n 2 | Necessary to remove forward and reverse from reads |
| –max-n | the maximum number of N’s in a trimmed sequence pair before they are discarded. |
Filtering out all trimmed pairs with one or more N’s allows counting primer matches in the R script below. For ITS data, we expect the target region to vary in length so we do not use -m and -l to force them to be a fixed length. If an ITS primer set suitable for merging the reads produced is used (as with BITS/B58S3), and we use the QIIME 2 dada2 plug-in to process the reads, we would set the truncation parameters to zero and filter the reads by quality instead.
The BITS/B58S3 primer set was devloped by Bokulich and Mills, 2013. For their test data the expected amplicon length was 183.6 ± 46.8 bp. Thus we can expect some read-through with most sequencing platforms.
Checking Primer Trimming
To follow along, download sequences for a few samples from NCBI project PRJNA377530. 10,000 sequences each are enough. Put them in the directory its_raw under your R project directory.
Trim the sequences using the cutadapt script above and place the trimmed sequence files in its_raw/trimmed under your R project directory.
Then run the following R code. You may download the code as its_trim_check.Rmd or copy and paste it into an Rmd file.
Load required libraries.
```{r, message=FALSE}
library(dada2)
packageVersion("dada2")
library(ShortRead)
packageVersion("ShortRead")
library(Biostrings)
packageVersion("Biostrings")
```
Put the untrimmed sequences in the directory raw_seqs.
Put the trimmed sequence files in the directory raw_seqs/trimmed.
```{r}
path <- paste0(getwd(),"/its_raw")
path
list.files(path)
```
Make separate lists of the forward and reverse sequence files sorted in the same order.
```{r}
fnFs <- sort(list.files(path, pattern = "_1.fastq", full.names = TRUE))
fnRs <- sort(list.files(path, pattern = "_2.fastq", full.names = TRUE))
fnFs
fnRs
```
Assign the forward and reverse primers to variables.
```{r}
# BITs forward primer
FWD <- "ACCTGCGGARGGATCA"
# B58S3 reverse primer
REV <- "GAGATCCRTTGYTRAAAGTT"
```
Calculate all orientaitons of the primers.
```{r}
allOrients <- function(primer) {
# Create all orientations of the input sequence
dna <- DNAString(primer) # The Biostrings works w/ DNAString objects rather than character vectors
orients <- c(Forward = dna, Complement = Biostrings::complement(dna), Reverse = Biostrings::reverse(dna),
RevComp = Biostrings::reverseComplement(dna))
return(sapply(orients, toString)) # Convert back to character vector
}
FWD.orients <- allOrients(FWD)
REV.orients <- allOrients(REV)
FWD.orients
REV.orients
```
Filter the sequences of all N's and put the results in sub-directory filtN. If N's are present in the sequences, it will inflate the number of matches found.
```{r}
fnFs.filtN <- file.path(path, "filtN", basename(fnFs)) # Put N-filtered files in filtN/ subdirectory
fnRs.filtN <- file.path(path, "filtN", basename(fnRs))
filterAndTrim(fnFs, fnFs.filtN, fnRs, fnRs.filtN, maxN = 0, multithread = FALSE)
```
Count how many times each orientation of each primer hits the forward and reverse sequences for the first fastq file.
```{r}
primerHits <- function(primer, fn) {
# Counts number of reads in which the primer is found
nhits <- vcountPattern(primer, sread(readFastq(fn)), fixed = FALSE) return(sum(nhits > 0))
}
FWdOrients.ForwardReads <- sapply(FWD.orients,
primerHits,
fn = fnFs.filtN[[1]])
FwdOrients.ReverseReads <- sapply(FWD.orients,
primerHits,
fn = fnRs.filtN[[1]])
RevOrients.ForwardReads <- sapply(REV.orients,
primerHits,
fn = fnFs.filtN[[1]])
RevOrients.ReverseReads <- sapply(REV.orients,
primerHits,
fn = fnRs.filtN[[1]])
rbind(FWdOrients.ForwardReads,
FwdOrients.ReverseReads,
RevOrients.ForwardReads,
RevOrients.ReverseReads)
```
Repeat for the trimmed sequences. I used cutadapt with parameters based on Callahan's R script and placed the trimmed sequence files in the sub-directory trimmed, i.e. under its_raw as in the next code block.
```{r}
path <- paste0(getwd(),"/its_raw/trimmed")
path
list.files(path)
```
Make lists of the forward and reverse sequence files.
```{r}
fnFs <- sort(list.files(path, pattern = "_R1.fastq", full.names = TRUE))
fnRs <- sort(list.files(path, pattern = "_R2.fastq", full.names = TRUE))
fnFs
fnRs
```
Tabulate matches of all orientations of the primers to the sequences in the first file.
```{r}
FWdOrients.ForwardReads <- sapply(FWD.orients,
primerHits,
fn = fnFs[[1]])
FwdOrients.ReverseReads <- sapply(FWD.orients,
primerHits,
fn = fnRs[[1]])
RevOrients.ForwardReads <- sapply(REV.orients,
primerHits,
fn = fnFs[[1]])
RevOrients.ReverseReads <- sapply(REV.orients,
primerHits,
fn = fnRs[[1]])
rbind(FWdOrients.ForwardReads,
FwdOrients.ReverseReads,
RevOrients.ForwardReads,
RevOrients.ReverseReads)
```
That's good, no hits to the trimmed sequences!
The script is meant to be run in R Studio. For the untrimmed sequences, you should get something like:
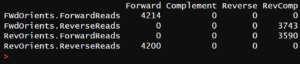
The numbers under the heading RevComp indicate a high degree of read-through. The reverse complement of the forward primer hit 3,743 of the reverse reads and the reverse complement of the reverse primer hit 3,590 of the forward reads.
And for the trimmed data:
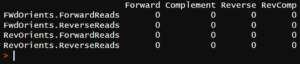
There are no hits for any orientation of either primer. Read-through eliminated!
Trimming Single (Unpaired) Reads
If you have single reads that contain the target region flanked by both primers, trim the reads using cutadapt’s linked adapter method. The reverse complement of the reverse primer is added to the forward primer sequence with three periods between the primer sequences.
Some technologies (PacBio, LoopSeq) return single reads. Or you may choose to merge first and trim second to eliminate overhangs. In any case, you can trim with a script similar to the following.
#! /bin/bash
# Trim forward and reverse primers from single reads.
# Input uncompressed files, output uncompressed files.
# The forward primer is 515f
fwd=GTGCCAGCMGCCGCGGTAA
# The reverse primer is 806rb
rev=GCACTACNVGGGTWTCTAAT
# The reverse complements of the reverse primer is:
rev_rc=ATTAGAWACCCBNGTAGTGC
cd ~/temp_seqs/ # Move into experiment level directory.
rm cutadapt_log.txt # Remove previous log file.
rm -rf trimmed # Remove previous results
mkdir trimmed # Make directory for the trimmed reads.
cd merged/ # Move into directory containing merged reads with primers present.
# Trim using cutadapt.
for f in $(ls *_merged.fastq)
do
cutadapt -a $fwd...$rev_rc \
--discard-untrimmed \
-e 0.11 -O 15 --max-n 0 \
-l 255 -m 250 \
-o ../trimmed/${f/_merged.fastq/_trimmed.fastq} \
$f >> ../cutadapt_log.txt
done
References
Apprill A, McNally S, Parsons R, Weber L. 2015. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat Microb Ecol 75:129-137.
Bokulich NA, Mills DA. 2013. Improved Selection of Internal Transcribed Spacer-Specific Primers Enables Quantitative, Ultra-High-Throughput Profiling of Fungal Communities. Appl Environ Microbiol 79:2519-2526.
Li B-B, Roley SS, Duncan DS, Guo J, Quensen JF, Yu H-Q, Tiedje JM. 2021. Long-term excess nitrogen fertilizer increases sensitivity of soil microbial community to seasonal change revealed by ecological network and metagenome analyses. Soil Biol Biochem 160:108349.