
Add a SeqMatch Database
SeqMatch preceded the RDP Classifier for as a means of classifying 16S rRNA gene sequences. It uses a kmer approach to finding the closest matches in a database. Here I provide instructions for adding two SeqMatch databases to RDPTools.
Click on this link to download the archive file SeqMatch_DBs.zip. If you installed RDPTools using the Docker image, place the file in the folder you mapped to the container (see creating-the-container in my GitBook instructions for installing RDPTools from a Docker image) and decompress it. Otherwise place it in your home directory or wherever you wish. Decompressing the file generates the directory SeqMatch_DBs with the following files:
SeqMatch_DBs release11_4_type_descriptions.txt release11_4_bac_isolate_descriptions.txt isolate_trainee_files release11_4_bac_isolates_1.trainee release11_4_bac_isolates_2.trainee release11_4_bac_isolates_3.trainee release11_4_bac_isolates_4.trainee release11_4_bac_isolates_5trainee release11_4_bac_isolates_6.trainee release11_4_bac_isolates_7.trainee release11_4_bac_isolates_8.trainee
As you can see, there are two databases; one for type strains and another for bacterial isolates.
Using SeqMatch
Entering the following command …
java -jar /usr/local/RDPTools/SequenceMatch.jar seqmatchgives a help message for the seqmatch program:
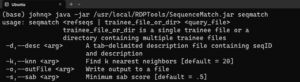
To classify sequences in a fasta file with the type strains database (edit the paths as appropriate), use the command:
java -jar /usr/local/RDPTools/SequenceMatch.jar seqmatch \
~/SeqMatch_DBs/release11_4_types.trainee \
query.fasta \
--desc ~/SeqMatch_DBs/release11_4_type_descriptions.txt \
--knn 20 \
--sab 0.5 \
--outFile query_classified.tsvTo classify sequences in a fasta file with the isolates database, use the command:
java -jar /usr/local/RDPTools/SequenceMatch.jar seqmatch \
~/SeqMatch_DBs/isolate_trainee_files \
query.fasta \
--desc ~/SeqMatch_DBs/release11_4_bac_isolate_descriptions.txt \
--knn 20 \
--sab 0.5 \
--outFile query_classified.tsv