
Read Characteristics
Q: Are the reads paired? How can you tell?
A: Yes, the reads are paired. The file names all end with either “_R1.fastq” or “_R2.fastq.”
Q: Do they require multiplexing, or are they binned by sample?
A: No. they do not require demultiplexing. Paired reads begin with the same prefix, in this case the sample name. The file names are in Zymo format so that they may be read with FIGARO.
Q: What are the lengths of these reads?
A: 301 bp.
Q: Are primers present?
A: Yes.
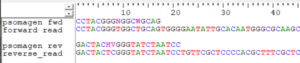
Q: What are the positions of the primers in the reads?
A: At the very ends of the reads as shown in the alignment below, so you may use anchored adapters to trim with cutadapt or the QIIME 2 cutadapt plugin.
FastQC/MultiQC (read quality)
Q: Are there samples that need to be removed because of low quality or too few reads?
A: No, there are more than 9,000 sequences in all fastq files. Forward read quality begins to fall below a Q value of 30 at about 260 bases and are above a Q value of 20 until the end at 301 bases. Reverse read quality begins to fall below a Q value of 30 at about 205 bases and below a Q value of 20 at about 255 bases. Depending on the expected length of the merged sequences, this may reduce the number of merged sequences.
Q: Are there Illumina adapter present?
A: Very few, all fastq files receive passing scores for Illumina adapter content. There is no need to remove Illumina adapters.
Decide on a Processing Strategy
There are several sub-questions to address in deciding a processing strategy. For example:
-
- Will you merge the forward and reverse reads?
- What merged length can be achieved?
- Will merging select against some taxa?
You may have to consult the literature, but DO NOT just follow others’ methods blindly. Check things out for your self. Do sanity checks as you go. Working with a small data set like this one lets you experiment with different methods and parameters quickly.
Determine lengths of the trimmed reads
First, determine the lengths of the trimmed reads. You will have to run cutadapt first and then get information on the lengths of the trimmed reads, e.g. with USEARCH -fastx_info. I found that after trimming, forward reads varied in length from 283 to 286 bp and the reverse reads varied in length from 279 to 285 bp.
Determine length of amplicon region
Next we need to determine the expected length of the target region. I made a Google search of the forward primer sequence and came up with the three references at the bottom of this page. Herlemann et al. (2011) designed the primers to sequences in the SILVA database and the primer names (Bact_341F and Bact_805R) give their positions on the Escherichia coli 16S rRNA gene. From the positions, the expected amplicon length including the primers is 805 -341 + 1 = 465, and 465 – 17 – 21 = 427 trimmed of the primers. Thijs et al. (2017) includes a table giving the amplicon length as 444 bp.
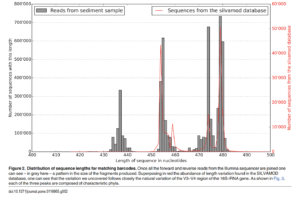
Another paper (Sinclair et al., 2015) included a figure (below) showing that the length distribution of the V3/V4 region varied and fell into three clusters between 433 and 482 bp. Sequences in the clusters, from left to right, were classified mainly to Archaea, Chloroflexi, and Bacteriodetes. The lengths in the figure include primer regions. Subtracting the sum of the primer lengths from 433 and 483 bp, we determine the expected amplicon length varies between 395 and 444 bp.
From the above, I decided to choose methods and parameters that allow obtaining 444 bp merged reads. The minimum length of the forward trimmed reads (283) plus the minimum length of the reverse trimmed read (279) minus the default overlap length for the plugin version of DADA2 (12) equals 550, so it is possible to merge at least some of the reads. The actual number of merged sequences obtained will depend on the read quality and the merging parameters chosen.
Perhaps, like me, your first thought is to process these sequences as you did the V4 region sequences. Let’s try that. Trim the primers. Because we want to use Figaro to determine truncation parameters, we need to make the trimmed sequences all the same length, say 270 bp.
#!/bin/bash
# Run cutadapt
# Activate qiime2 before running this script
base_dir=~/psomagen_exercise
cd $base_dir
seq_dir=$base_dir/raw_sequences
# Psomagen primers are
fwd=CCTACGGGNGGCWGCAG
rev=GACTACHVGGGTATCTAATCC
cd $base_dir
rm cutadapt.log
rm -rf trimmed
mkdir trimmed
cd $seq_dir
for f in $(ls *.fastq);
do
cutadapt -g $fwd -G $rev \
--discard-untrimmed \
--match-read-wildcards \
-e 0.1 -O 15 -m 270 -l 270 -j 4 \
-o ../trimmed/${f/_16s_R1.fastq/_trimmed_R1.fastq} \
-p ../trimmed/${f/_16s_R1.fastq/_trimmed_R2.fastq} \
$f ${f/_R1.fastq/_R2.fastq} >> $base_dir/cutadapt.log
doneThen run Figaro on the trimmed sequences, specifying an amplicon length of 444 bp.
python figaro.py -i ~/psomagen_exercise/trimmed/ -o ~/psomagen_exercise/trimmed/ -f 1 -r 1 -a 444 -F zymoI found the top Figaro result to be:
{"trimPosition": [258, 208], "maxExpectedError": [4, 3], "readRetentionPercent": 77.56, "score": 64.56325970770467}With these parameters, run the QIIME2 script through the DADA2 step and output the denoised.qzv file so that we can see how many sequences were retained at each step.
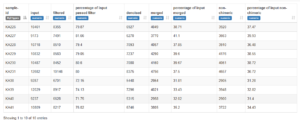
Retention is not very good. Many sequences are lost during merging. That is because I left the max_merge_mismatch at the default value of 0.
Experimenting with USEARCH -fastq_mergepairs, I learned that the mean overlap length is ~124 bp. Requiring no mismatches over so long a length is simply too strict. I had to allow 12 mismatches to get ~80% of the read pairs to merge. So I tried different values for max_merge_mismatch. A value of 5 gave the results below and a value of 10 did no better. So I chose 5 to process the full data set. I did not expect any overhangs, and there were very few, possibly a result of PCR error. I included the option to remove overhangs.
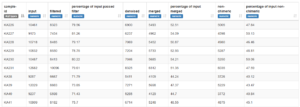
Not fantastic, but better.
The transition error plot looks very good.
![]()
References
Herlemann DPR, Labrenz M, Juergens K, Bertilsson S, Waniek JJ, Andersson AF. 2011. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. Isme Journal 5:1571-1579.
Sinclair L, Osman OA, Bertilsson S, Eiler A. 2015. Microbial Community Composition and Diversity via 16S rRNA Gene Amplicons: Evaluating the Illumina Platform. PLOS ONE 10:e0116955.
Thijs, S., Op De Beeck, M., Beckers, B., Truyens, S., Stevens, V., Van Hamme, J. D., & Vangronsveld, J. 2017. Comparative evaluation of four bacteria-specific primer pairs for 16S rRNA gene surveys. Frontiers in microbiology, 8, 494.