
Read Characteristics
Q: Are these paired reads?
A: Yes.
Q: If paired, are they interleaved or are forward and reverse reads in separate files?
A: These are paired reads. The forward and reverse reads are in separate files.
Q: Do they require binning by sample?
A: No, the first 3 characters of the file names are the sample names.
Q: How long are the reads?
A: Running usearch11 -fastx_info tells us that the reads are 250 bp.
Q: Are the quality scores binned?
A: Yes, there are only 3 characters in quality score lines of the fastq files: comma, semi-colon and upper case F. These decode to quality scores of 11, 25 and 37. An uncalled base woudl be 2. The sequences should not be processed with the QIIME2 version of DADA2.
Q: Align the primers with the reads. Where do the primers hit? At the very ends of the reads, or is there some padding prior to the primer sequence? How long are the primers?
A: To answer these questions, make fasta files of one forward and one reverse read. Catenate these with the primers and import them into BioEdit or Mega11. Then you can align the primers with their respective sequences to get something like this:

The forward primer is 16 nucleotides long and hits the forward read beginning at the 7th position.
The reverse primer is 20 nucleotides long and hits the reverse read beginning at the 7th position .
Thus, after primer trimming the forward reads will be 250 – 6 -17 = 227 bases and the reverse reads will be 250 – 6 – 20 = 224 bases.
Q: How long is the target region?
A: The primers target the V3/V4 region. You can determine this by conducting a Google search of the primers or by reading the Novogene’s documents. From the literature the target region is 440 bp, the same as for the Psomagen example. If we merge the trimmed sequences with usearch -fastq_mergepairs the mean merge length is 418.52 base and the mean alignment length is 32.48. For usearch, the default minimum over lap length is 16. If we set this to 12 instead with the fastq_minovlen parameter, the stats are the same.
Q: Assuming an overlap length of 12 bp for merging, what is the maximum length of merged reads?
A: 227 + 224 – 12 = 439. There is some danger of excluding taxa with longer V3/V4 sequences, especially as some truncation for quality will likely be necessary.
Read Quality
Q: Run fastqc and multiqc. Are there any samples that should be removed for poor quality or too few reads?
A: No, all are excellent.
Q: Are there warnings about the presence of Illumina adapters? If so, they should be removed before proceeding.
A: No, Illumina adapters are not present.
Process the Reads
Decide on a processing strategy.
Run Figaro on the raw reads using forward primer length of 17 + 6 = 23 and reverse primer length of 20 + 6 = 26.
Q: Will this work if the amplicon length is 444?
A: No, you get the message/error “Combined read lengths are less than the required combined length.”
Because the quality scores are binned, I decided to process the reads in R using DADA2. Presently (30 March 2026) the latest QIIME2 DADA2 version does not inclulde an option for hadling binned quality scores, but the R version does. The part of the R code for learning errors from continuous quality scores is:
errF <- learnErrors(filtFs, multithread=TRUE)
errR <- learnErrors(filtRs, multithread=TRUE)This results in transition error plot with large unacceptable downward loops between Q = 25 and Q = 37 as in the plot below.
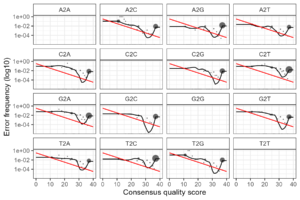
Teh poionts are scaled by their relative abundance, so we can see that most of the data is at Q 37.
Callahan has included a function to generate a learn errors function given a vector of the binned quality scores. The part of the R code for learning errors taking the binning into account is:
novaseqBinnedErrFun <- makeBinnedQualErrfun(c(2, 11, 25, 37))
errF <- learnErrors(filtFs, multithread=TRUE, errorEstimationFunction = novaseqBinnedErrFun)
errR <- learnErrors(filtRs, multithread=TRUE, errorEstimationFunction = novaseqBinnedErrFun)Using this method of learning the transition error rates results in much more acceptable transition error plots as below.
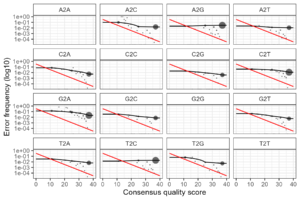
The read quality is exceptional. So rather than using Figaro determined truncation parameters, try just filtering on quality and change the overlap length to 10 (from the default of 12).
Q: What is the overall read retention?
Allowing for the binned quality scores and decreasing the required overlap length from 12 to 10 resulted in retention as in the following table:
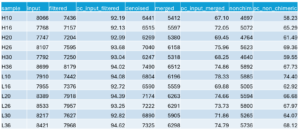
You may download an Rmd version of the R script I used from here.
Q: What is the length distribution of the representative sequences?
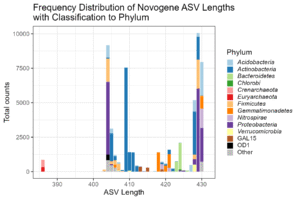
The few short sequences at 386 bp are Archaea. There are no ASVs over 430 bp. If species with greater sequence length were present we would miss them.
I think it is a mistake to sequence the V3/V4 region with 2 X 250 bp chemistry as was done here. The reads are not long enough to capture the full expected length of the amplicons. Be careful in choosing your sequencing method.