RDP Classifier
To obtain the most recent version of the RDP Classifier, follow the installation instructions on Installing the Stand-alone RDP Classifier. The RDP Classifier within the Docker distribution of RDPTools is an earlier version. Usage instructions can be found here.
Besides simply classifying sequences, the RDP Classifier can perform a supervised analysis of community data. If one gives it a list of sample fasta files and requests output in the hierarchical format, the results can be imported into phyloseq as an experiment level object containing both an OTU table and a classification table.
Four databases are included with the RDP Classifier as shown in the table below. These are chosen with the --gene parameter. The default is 16srrna for classifiying bacteria and archaea. The others are for classifying fungi – fungallsu by 28S rRNA gene sequences and the fungalits_warcup and fungalits_unite by the “full length” ITS sequence. The ITS training data included partial SSU sequence, ITS1, 5.8S, ITS2 and partial LSU sequence. Thus, they can be used to classify either ITS1 or ITS2 amplicons.
| Database | Last updated |
| 16srrna | 2023 |
| fungallsu | 2014 |
| fungalits_warcup | 2016 |
| fungalits_unite | 2014 |
The RDP Classifier can be trained on other/more recent datasets if the taxonomy tags are formatted appropriately and represent a valid phylogeny (meaning the same rank cannot occur in more than one higher rank). See Training the RDP Classifier for an example of how I trained the RDP Classifier on UNITE data.
USEARCH sintax
The USEARCH sintax (simple non-Bayesian taxonomy classifier) function was launched in 2016 as an alternative to the RDP Bayesian classifier. It predicts taxonomy by kmer similarity to sequences in a reference database and being non-Bayesian does not require training.
In June 2024 Robert Edgar donated the USEARCH binaries to the public domain, so you may now download the 64-bit versions for your operating system from GitHub. To install, simply place the file in a directory on your path, but I suggest that you shorten the name to simply usearch11.
The taxonomy is taken from the reference sequence descriptions, so these descriptions must be correctly formatted. Reference sequences formatted for use with sintax are available from https://www.drive5.com/usearch/manual/sintax_downloads.html.
You may build a database from the reference sequences if you wish, but it is not required. It does make the classification run more quickly. For example, to build a reference database file from the RDP Classifier training set version 18, enter:
wget https://drive5.com/sintax/rdp_16s_v18.fa.gz --no-check-certificate
gunzip rdp_16s_v18.fa.gz
sed -i 's/TALKALIHALOBACILLUS//' rdp_16s_v18.fa
usearch11 -makeudb_usearch rdp_16s_v18.fa -output sintax_rdp_16s_v18.udbWhen I tried building a database from rdp_16s_v18.fa with the VSEARCH –makeudb_usearch command (see below) it reported WARNING: 8 invalid characters stripped from FASTA file: I(2) L(5) O(1). This was because one of the sequences began with TALKALIHALOBACILLUS. That is why I used the third line to correct this error by removing the string TALKALIHALOBACILLUS from the file.
To classify representative (unknown) sequences, enter the appropriate line below, depending on whether or not you are using a database file. Both fastq and fasta files are accepted as input.
usearch11 -sintax unknown_seqs.fastq -db rdp_16s_v18.fa -strand both -sintax_cutoff 0.8\
-tabbedout sintax_classified.tsv
usearch11 -sintax unknown_seqs.fastq -db sintax_rdp_16s_v18.udb -strand both -sintax_cutoff 0.8 \
-tabbedout sintax_classified.tsv VSEARCH sintax
VSEARCH is part of QIIME 2 but there is no plug-in interface to the VSEARCH sintax command. However, with an activated QIIME 2 environment the command VSEARCH --sintax can be run natively . The commands are the same as for USEARCH sintax except that two hyphens must preceed the arguments:
wget https://drive5.com/sintax/rdp_16s_v18.fa.gz --no-check-certificate gunzip rdp_16s_v18.fa.gz sed -i 's/TALKALIHALOBACILLUS//' rdp_16s_v18.favsearch --makeudb_usearchrdp_16s_v18.fa -output vsearch_sintax_rdp_16s_v18.udb vsearch --sintax seqs_2_classify.fasta --db vsearch_sintax_rdp_16s_v18.udb --strand both \ --sintax_cutoff 0.8 --tabbedout vsearch_classified.tsv
QIIME 2 Classifiers
The q2-feature-classifier plug-in supports three methods of classification:
-
-
- classify-concensus-blast
- classify-concensus-vsearch
- classify-sklearn
-
The first two are alignment based methods that find a consensus taxonomy assignment from N top hits. I do not recommend either of these and do not cover them here. Better performing overall is the classify-sklearn method with a Naive Bayes classifier. These require training which is computationally intensive so can take a long time, but training has to be done only once (for a particular version of QIIME 2). And there are pre-built classifiers at for 16S here and for ITS here. The classifiers are specific to a version of QIIME 2 – the version of scikit-learn used to build the classifier and in the QIIME 2 version being used to classify the sequences must match. For 16S, the classifier should be trained on the sequences trimmed to the variable region under study. For ITS, it is recommended that the classifier be trained on full-length sequences.
Instructions for building classifiers can be found here and here. Be prepared for frustration. Not all of the links to resources work.
The following code serves as an example of using the QIIME2 Baysian classifier trained on Greengenes 2 V4 sequences to classify a few (50) V4 representative sequences.
#!/bin/bash
# Classify with QIIME2 version 2024.5
# Download pre-built Greengenes2 V4 classifier
wget https://data.qiime2.org/classifiers/sklearn-1.4.2/greengenes2/2022.10.backbone.v4.nb.sklearn-1.4.2.qza
# Download a few V4 sequences
wget https://john-quensen.com/wp-content/uploads/2024/11/v4_rep_seqs.zip
unzip v4_rep_seqs.zip
# Import sequences to be classified
qiime tools import \
--type 'FeatureData[Sequence]' \
--input-path v4_rep_seqs.fasta \
--output-path rep-seqs.qza
# Classify
qiime feature-classifier classify-sklearn \
--i-classifier 2022.10.backbone.v4.nb.sklearn-1.4.2.qza \
--i-reads rep-seqs.qza \
--o-classification taxonomy.qza
# Export
qiime tools export \
--input-path taxonomy.qza \
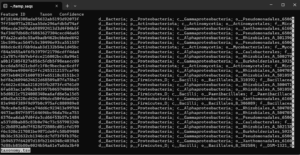
--output-path ./The result is written to the file taxonomy.tsv. If you run the above example and view the file with less, you should see something like: