The first step in working with sequencing data should always be evaluating its quality. This can be done with the FastQC program which generates html formatted reports you can read in your browser, but if you have hundreds of paired-read files, do you really want to examine them all one by one? I know I don’t, and so up until now I have been examining a subset, say 10 or 12 of the files. But I recently discovered an R package capable of summarizing the results, and using tidyverse functions you can filter the summary to show only samples with potential problems.
The name of the package is, unsurprisingly perhaps, fastqcr and it can be installed from the CRAN repository using the “Packages” tab in RStudio or by entering the following command in the R terminal:
install.packages("fastqcr")
See the GitHub page https://github.com/kassambara/fastqcr for full instructions on the use of the package.
Not all of the reports are pertinent for processing 16S rRNA gene amplicon data, so I have been using something like the following to examine such data:
library(fastqcr)
library(dplyr)
rpt <- qc_aggregate(qc.dir = "fastqc/")
rpt |> print(n=11)
Where my FastQC results are in the directory fastqc/ under my working directory. This produces something like:
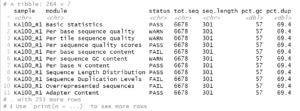
From this we can see the names of the individual reports (modules) per file and that the sequences should all be 301 bp in length. With this information we can filter rpt to show which samples have potential problems regarding overall sequence quality and the presence of uncalled bases or adapter sequences.
rpt |>
dplyr::filter(module %in% c('Per base sequence quality',
'Per base N content',
'Adapter Content'),
status != 'PASS' | `seq.length` != 301) |>
print(n=Inf)
For this example data, we get the following:
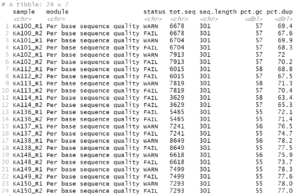
If there were no potential problems for the items, you would get an empty result.

