If FastQC warns that some of your sequences contain Illumina adapters, you can remove those sequences with bbduk.sh from the bbmap tool. Bbmap is a java program and is partof the bbtools suite of programs written by Brian Bushnell of JGI. It can be installed in several ways.
Installation Option 1
Because it is written in java, it can be run on any platform that has java installed. Download the latest version of the program from Sourceforge to your home directory and decompress it with tar (tar xzf file_name.tar.gz). If you use this option, to run bbduk.sh you must give the complete path to the shell script.
Installation Option 2
Alternatively, you can install the program using an environment manager, e.g. micromamba.
micromamba create --name bbmap -c bioconda bbmapInstallation Option 3
And if you are using a Mac computer, you may install the bbtools suite with Homebrew:
brew install bbtoolsTo use the tool, you will also need a database of all of the possible adapter sequences to search against. Download them as a fasta file from here or here.
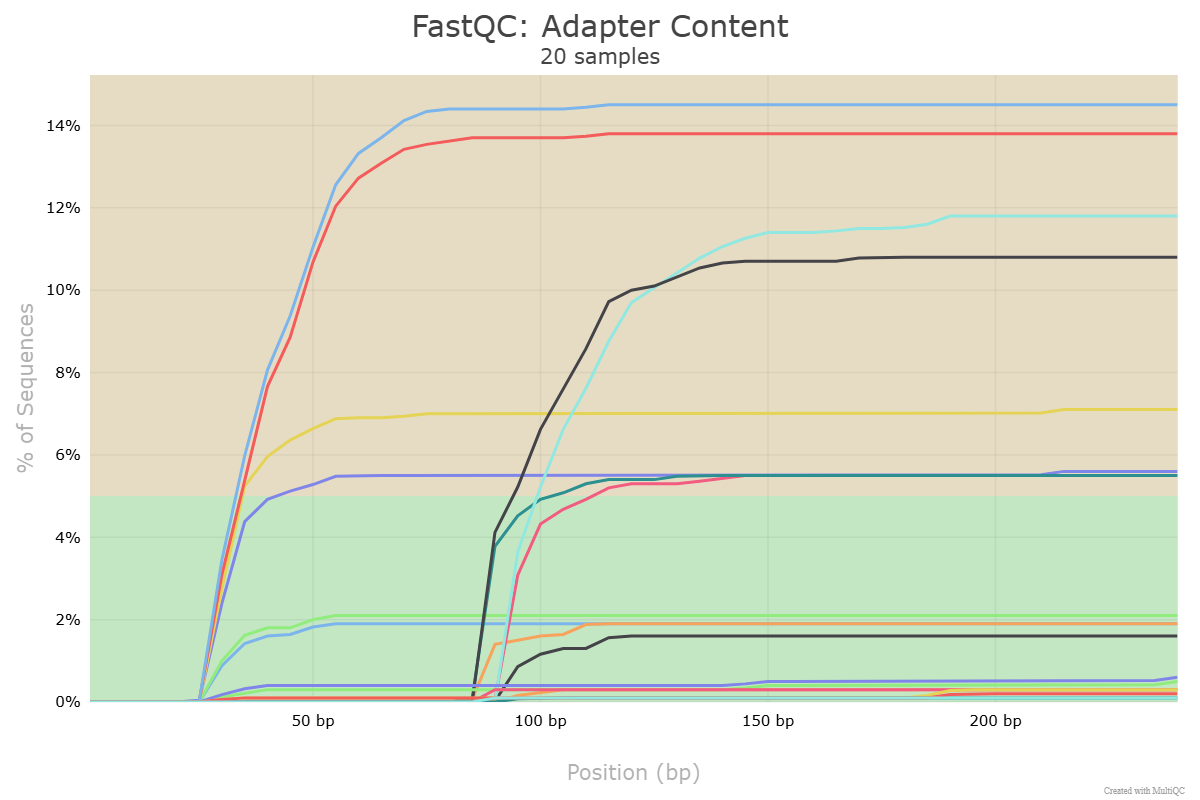
I have provided some sequences for you to use to test your bbmap installation. If you run FastQC and MultiQC on these files, among the MultiQC reports you should get one like this:
Test you installation by running following script (first activating the bbduk environment if necessary) from the directory containing the sequences. Remember that if you used installation Option 1 above, you must provide the path to bbduk.sh in the command. In my case, this is ~/bbmap/.
#!/bin/bash
rm bbduk_log.txt
rm -rf bb_out
mkdir bb_out
for f in $(ls *_1.fastq); do
#~/bbmap/ # My path to bbduk.sh
bbduk.sh in1=$f in2=${f/_1.fastq/_2.fastq} out1=bb_out/$f out2=bb_out/${f/_1.fastq/_2.fastq} \
ref=bb_adapters.fa k=17 mink=7 ktrim=rl hdist=1 qtrim=r trimq=20 minlen=100 tpe tbo \
2>> bbduk_log.txt
doneThe filtered sequences are written to the directory bb_out. If you run FastQC and MultiQC on the filtered sequences, you should not find any adapter sequences: