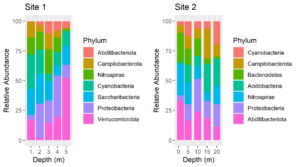
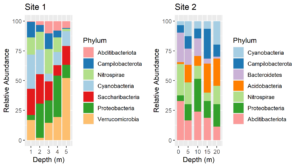
After processing amplicon sequencing data with QIIME2, I like to import QIIME2 results into R as an experiment level phyloseq object. Such phyloseq objects have slots for an OTU or ASV table, a taxonomy table, representative sequences, a tree file of the representative sequences and a sample data table. Thus, all of the components can be saved together in a single file. The QIIME2 scripts on this website include outputting QIIME2 artifacts into files that are easily imported into phyloseq. Below is an R script for importing all of the components into an experiment level phyloseq object.
If necessary, install the phyloseq and Biostrings packages from Bioconductor. The speedytax package is installed from CRAN.
library(phyloseq)
library(Biostrings)
library(speedytax)
# Read in the otu/asv table
asv <- read.table("otu_table.tsv", header = TRUE, row.names = 1, sep = "\t")
# Convert the otu/asv table into a phyloseq object.
asv <- phyloseq::otu_table(asv, taxa_are_rows = TRUE)
# Read in the sample data table; row names must match otu/asv table sample names.
sam <- read.table(file = "sample_data.tsv", header = TRUE, row.names = 1, sep = "\t")
# Convert the sample data into a phyloseq object
sam <- phyloseq::sample_data(sam)
# Read in the fasta file of representative sequences.
rep_seqs <- Biostrings::readDNAStringSet("dna-sequences.fasta", format = "fasta")
# Read in the classification table. In this example the representative sequences
# were classified with the RDP Classifier. speedytax includes other functions for
# importing QIIME2 and USEARCH sintax classification files.
rdp_taxa <- speedytax::import_rdp_tax_table("rdp_214_classified.txt", confidence = 0.5)
# Read in the tree file.
my_tree <- phyloseq::read_tree("rooted_tree.nwk")
# Package all into a single experiment level phyloseq object.
expt <- phyloseq::phyloseq(asv, sam, rep_seqs, rdp_taxa, my_tree)
expt
An often performed subsequent step is to filter the results taxonomically, for example removing taxa/representative sequences that are not classified or that may be chloroplast sequences. In the example above, the representative sequences were classified with the RDP Classifier, in which case I filter the results with the following R script.
# Copy expt to expt_filt
expt_filt <- expt
# Filter taxonomically.
# Get the rank names.
rank_names(expt_filt)
# Check classification at the domain level.
get_taxa_unique(expt_filt, taxonomic.rank = "Domain")
# Keep only taxa/sequences classified as Bacteria or Archaea.
expt_filt <- subset_taxa(expt_filt, Domain %in% c("Bacteria", "Archaea"))
# Re-check classification at the Domain level.
get_taxa_unique(expt_filt, taxonomic.rank = "Domain")
# Check classification at the phylum level
get_taxa_unique(expt_filt, taxonomic.rank = "Phylum")
# Remove taxa/sequences not classified to the phylum level
expt_filt <- subset_taxa(expt_filt, Phylum != "uncl_Bacteria")
expt_filt <- subset_taxa(expt_filt, Phylum != "uncl_Archaea")
# Re-check classification at the phylum level
get_taxa_unique(expt_filt, taxonomic.rank = "Phylum")
# Subset to taxa/sequences classified to Cyanobacteria.
cyan <- subset_taxa(expt_filt, Phylum == "Cyanobacteriota")
# Check classification of Cyanobacteriota at the class level
get_taxa_unique(cyan, taxonomic.rank = "Class")
# Remove taxa/sequences classified to chloroplasts
# or potentially to chloroplasts.
expt_filt <- subset_taxa(expt_filt, Class != "Chloroplast")
expt_filt <- subset_taxa(expt_filt, Class != "uncl_Cyanobacteriota")
# Check tht potential chloroplast taxa/sequences have been removed.
cyan <- subset_taxa(expt_filt, Phylum == "Cyanobacteriota")
get_taxa_unique(cyan, taxonomic.rank = "Class")
# Print summary of the experiment level phyloseq object
expt_filt
# Save the taxonomically filtered experiment level object.
saveRDS(expt_filt, file = "expt_filt.rds")
A similar procedure can be applied when another classifier is used to classify the representative sequences. For example, potential chloroplast sequences will not be found if chloroplast sequences are not part of the database used to build the classifier.